UV light from the sun is just one of the many factors we are exposed to on a daily basis that have the potential to damage our DNA. These breaks are efficiently repaired in healthy cells by distinct epigenetic and molecular machinery. However, if the damage is overwhelming or the repairs are imperfect, genomic mutations arise.
Aggressive tumours often carry defects in their ability to repair DNA. Therefore, these cancer cells have a high mutation rate that allows them to quickly overcome the immense selective pressures of the human body. However, recent discoveries about the epigenetic process behind this evolutionary advantage are now enabling modern medicine to turn it into a weakness.
Tumour Initiation
Most mutations won’t have any noticeable effect unless they occur in genes promoting or protecting against cancer, or in the non-coding elements regulating them. Such mutations, termed driver mutations, give cells rebellious intents, allowing them to grow faster, ignore their neighbours and appropriate the surrounding cellular space by becoming a tumour. In these early stages, our immune system, coupled with cellular intrinsic and extrinsic tumour suppressor mechanisms, will be quick to prevent the expansion of mutated cells. Therefore, many young tumours will not survive long enough to be noticed. However, the tumours that overcome these obstacles demonstrate that the key to cancer’s growth and spread is rapid evolution.
Tumour Evolution
Multiple driver mutations disrupt DNA repair machinery. They can affect the proteins that actively repair the DNA themselves, or the epigenetic effectors that regulate them. In either case, such mutations promote a tremendous mutation rate, drastically increasing the probability of generating novel driver mutations. As new advantageous mutations accumulate, cancer cells rapidly adapt and thrive in the highly selective and everchanging environment they occupy.
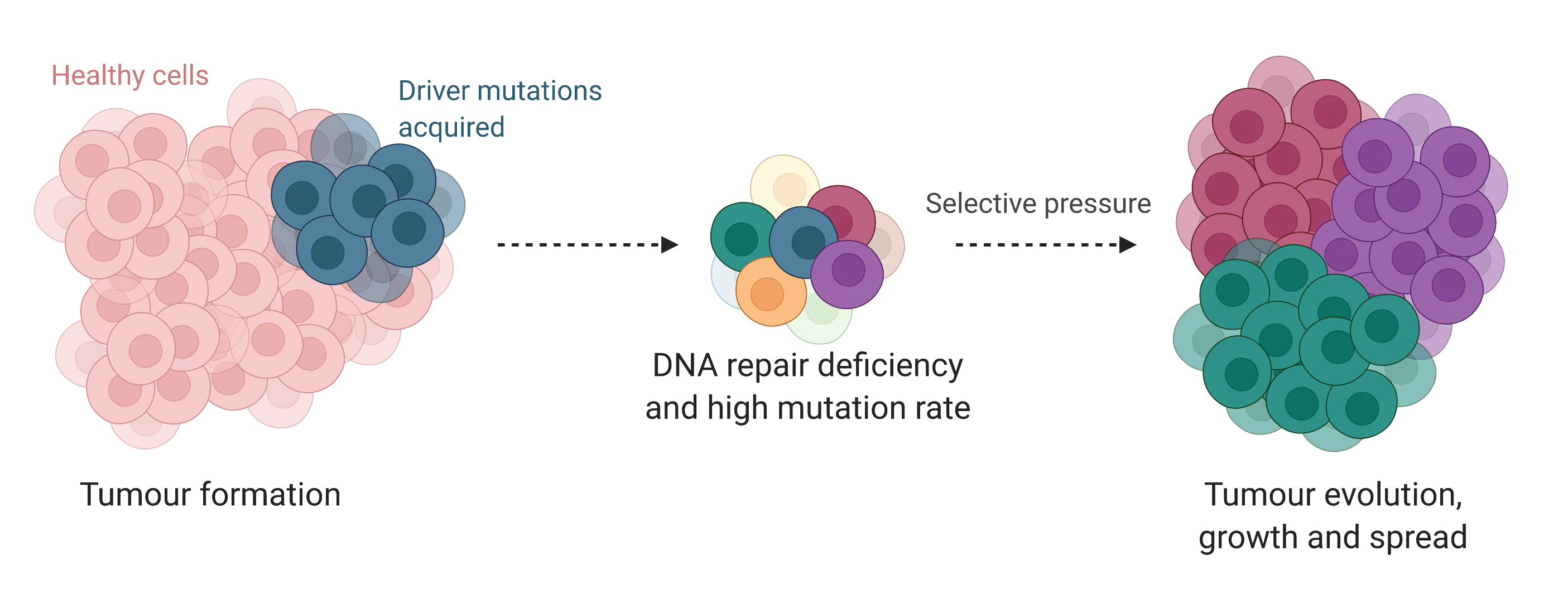
However, the evolutionary advantage of highly mutating cancers comes at a cost. Indeed, they rely heavily on the complementary DNA repair machinery they still possess to maintain a minimal level of DNA repair sufficient to survive and proliferate massively. And it turns out, their impaired DNA repair mechanisms may be one of their greatest weaknesses.
Cancer’s Achilles’ Heel?
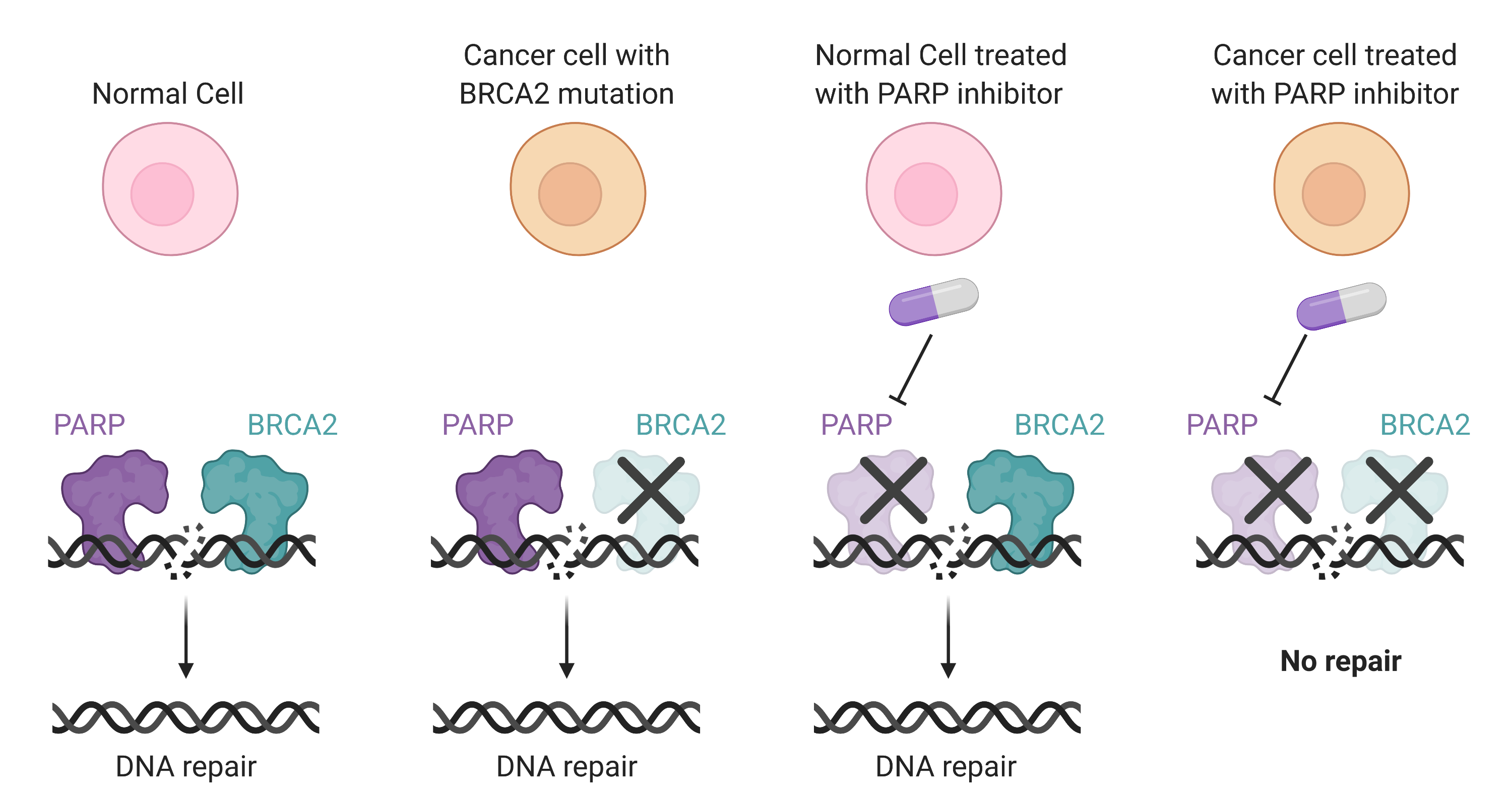
After years of studying DNA repair mechanisms, modern medicine now has the means to launch a targeted strike on rapidly mutating tumours, such as those harbouring the common BRCA2 mutations.
In these cancers, PARP1 is necessary to maintain a minimal level of genomic integrity. PARP1 acts as an epigenetic foreman on DNA break sites. It marks the DNA-associated proteins around the sites of breakage to recruit and organize the proteins repairing the break, independently of BRCA2. Without the coordination of DNA repair by PARP1, DNA damage can be repaired in BRCA2-dependant manner. But if both PARP1 and BRCA2 are missing, cells are overwhelmed by DNA damage and they cannot survive. Such a fatal relationship between two proteins or pathways is termed synthetic lethality.
Following this discovery, researchers and oncologists have developed pharmaceuticals targeting PARP1’s activity. These PARP inhibitors do not severely affect normal cells, as their intact DNA repair machinery can overcome the loss of PARP1. However, in cancer cells with a highly unstable genome, PARP inhibitors will result in lethal damage to their DNA.
PARP inhibitors have already shown great efficacy in the clinic for some specific cancers. But, PARP1 is not the only targetable epigenetic foreman coordinating DNA repair. Therefore, current research is aimed at identifying novel synthetic lethality between specific driver mutations and therapeutically targetable epigenetic regulators of DNA repair. There is hope that these new therapeutic approaches will create an insurmountable selective pressure driving highly mutating tumours to the verge of extinction.