Epigenetic Control of the Epithelial to Mesenchymal Transition in Cancer Metastasis
Epithelial to mesenchymal transition (EMT) is at the heart of cancer metastasis. Abnormal histone post-translational modifications affect expression of key EMT marker genes, potentiating this process. This article briefly introduces metastasis and EMT and describes how epigenetics plays a role in cancer progression.
Cancer Metastasis
Metastasis is the process where cancer cells from a primary site spread to secondary sites. Metastasising cancers have a significantly poorer prognosis than non metastasising cancers. Hence, a lot of oncology research focuses on understanding the biology of metastasis and its underlying causes.
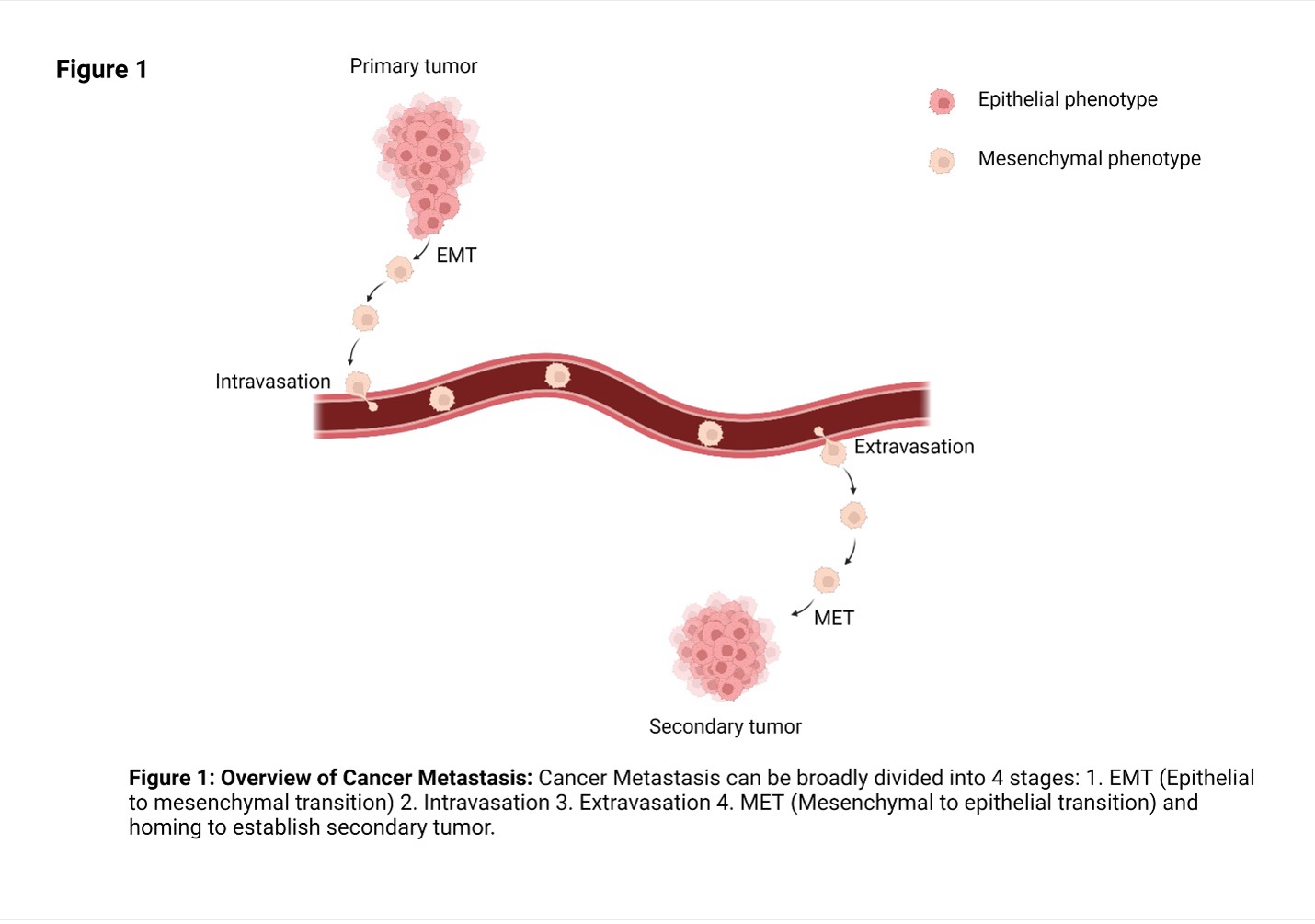
Metastasis can be broadly divided into the following steps:
1. A group of abnormal cells are found at the primary tumor site which have not yet invaded the surrounding tissue. This condition is called carcinoma in situ.
2. Some cancer cells in the primary tumor acquire invasive and migratory phenotype. One centrally important process for acquiring such phenotype is the cell biological process called epithelial to mesenchymal transition (EMT).
3. Migratory cancer cells enter the circulation through blood or lymph vessels.
4. Migratory cancer cells exit blood/lymph vessels by extravasation through vascular walls at a distant tissue or secondary site.
5. At the secondary site, cancer cells reacquire their original nonmigratory phenotype through mesenchymal to epithelial (MET) transition and establish microscopic colonies.
6. Microscopic colonies proliferate to form clinically detectable metastatic lesions.
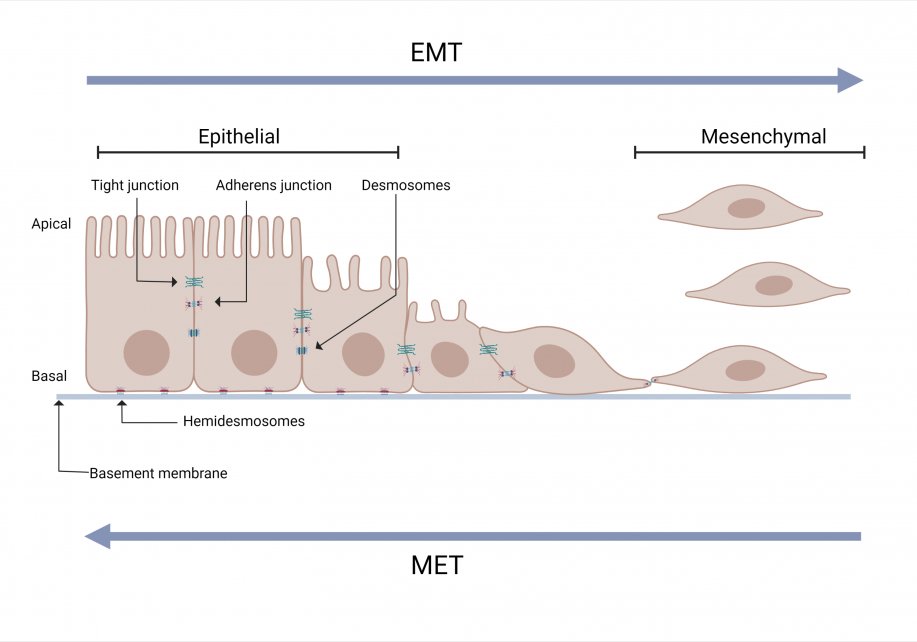
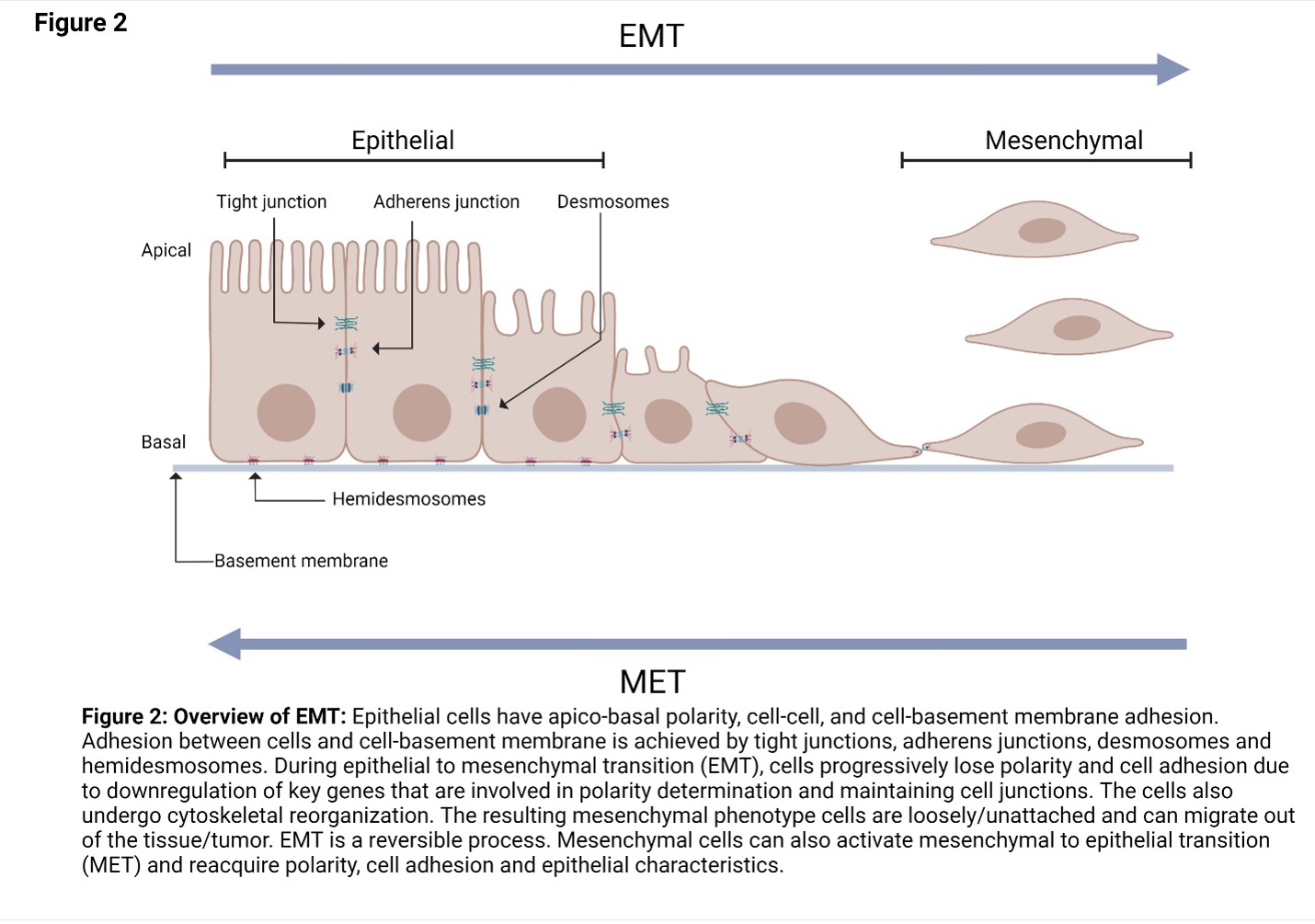
Epithelial to Mesenchymal Transition (EMT)
EMT is a normal biological process that occurs during embryogenesis, inflammation, wound healing and fibrosis. For example, during embryonic development, EMT is required for the establishment of the germ layers endoderm, mesoderm, and ectoderm.
EMT is induced by a range of factors including hypoxia, cytokine, growth factors, immune responses to the tumor and anticancer drug treatment. Due to EMT, some cancer cells in the primary tumor lose cell-cell or cell-basement membrane adhesions and undergo cytoskeletal reorganization.The cells transition from an epithelial phenotype to hybrid epithelial-mesenchymal or mesenchymal phenotype. These cells can migrate out of the primary tumor.
Intravasation and Extravasation
The migratory metastatic cells next invade the nearest lymph or blood vessel and gain access to circulation by a process called intravasation. Intravasation involves complex interactions between the cancer cells and the vascular endothelial cells.
Malignant cells in circulation are carried to the secondary sites of metastasis. At the secondary site, the cancer cells exit from the blood/lymph vessel by interacting with endothelial cells and this process is called extravasation (Figure 4).
Cells that have extravasated undergo mesenchymal to epithelial transition (MET). During MET, the cells re-acquire epithelial phenotype. Cell-adhesions are re-established, and a secondary tumor is formed from the metastatic cells. This ‘homing’ process is highly specific and depends also on the tissue microenvironment.
EMT and Histone Modification
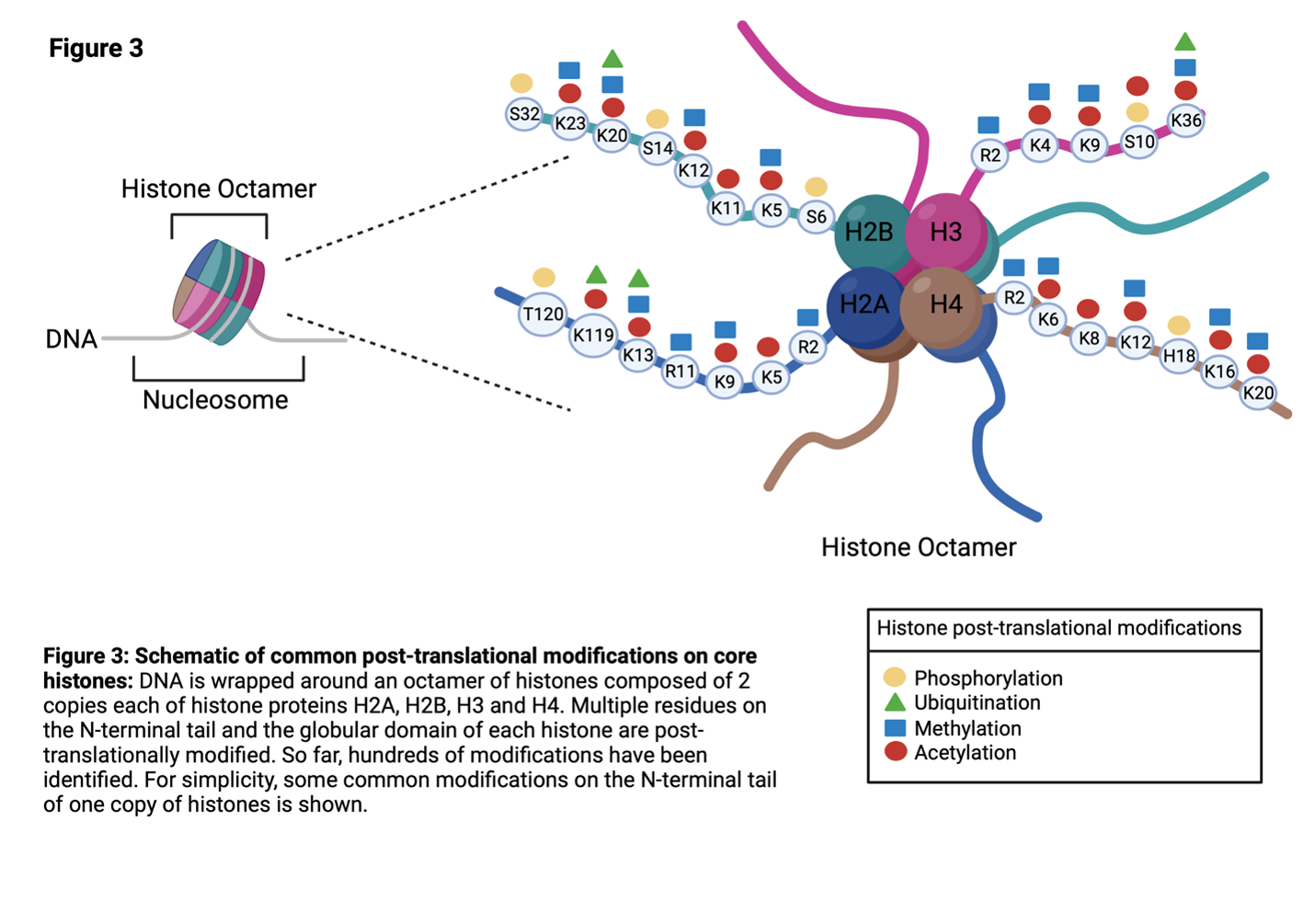
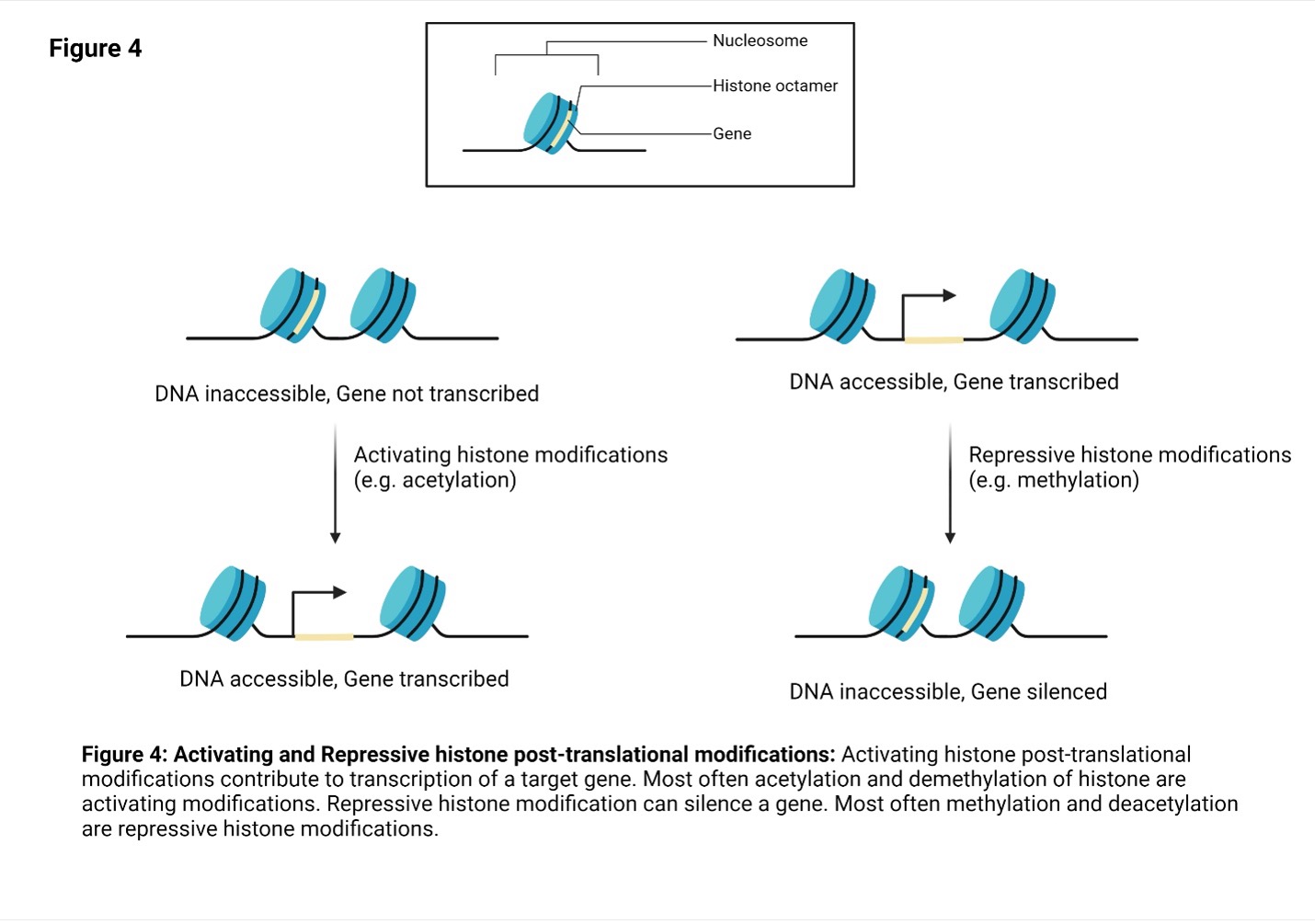
Histones are proteins that play a role in packaging DNA into chromatin. In the nucleus of cells, DNA is wrapped around an octamer of histones composed of 2 copies each of histones H2A, H2B, H3 and H416 (Figure 4). The composition of the octamer is not set in stone, and H2A, H2B, H3 and H4 are sometimes replaced by histone variants.
The histone proteins have multiple residues mainly on the N-terminal tail region that can be post translationally modified (Figure 3). Importantly, post-translational modification of the histone and the residue can have varying consequences including changes in the accessibility of DNA to transcription machinery. Since this can affect gene expression, enzymes responsible for histone modifications are tightly controlled. Aberrant expression of these enzymes are linked to many diseases including cancer.
Depending on their effect on gene expression, there are two broad categories of histone modifications: activating and repressive (Figure 4). Activating histone modifications include acetylation, demethylation and, rarely, methylation. Histone acetyl transferases and histone demethylases are responsible for these modifications. Repressive histone modifications include methylation, deacetylation and, rarely, demethylation. Histone methyl transferases and deacetylases are responsible for these modifications.
The main way histone modifications play a role in regulating EMT is by activation of epithelial gene expression and/or repression of genes responsible for mesenchymal-like phenotype. In metastasising cancers, these regulations are attenuated. Epigenetic research has led to a better understanding of the specific genes involved in this process.
Activating histone modifications
Wang et. al. showed that hypoxia induced EMT leads to significant increase in activating histone modification- acetylation of histone 3 lysine 4 at the promoter of genes GLI1 and SMO22. GLI1 and SMO over-expression leads to significantly increased migration and invasion capabilities of cancer cells as shown in head and neck squamous cell carcinoma.
Snail (SNAI1) and Slug (SNAI2) are two major transcription factors that promote EMT in both development and cancer contexts. The growth factor TGF-beta has been shown to be highly expressed in metastatic breast cancer. It has been shown that, Snail/Slug overexpression leads to enrichment of activating histone modification-histone 3 lysine 9 acetylation at the promoter of TGF-beta pathway protein TGF beta receptor 2 (TGFBR2) leading to overexpression of TGFBR223. Histone acetyl transferase hMOF has been shown to maintain expression of genes required for epithelial phenotype, such as TMS1, CDH1 and ESR1, by acetylation of histone 4 lysine 16 at the promoter. In some breast cancer and medulloblastoma, hMOF is down regulated.
Demethylation of the repressive histone methyl mark at the promoter of genes that induce EMT has also been associated with cancer metastasis. Histone demethylase JMJD2B has been shown to remove histone 3 lysine 9 methylation from the promoter of the vimentin (VIM) gene leading to its activation. Along with other genes, vimentin plays a key role in EMT. Histone demethylase PHF8 activates EMT inducer genes ITGB2, ITGAM and ITGA9 by demethylation of histone 3 lysine 9 at the promoter. Growth factor TGF-beta induces EMT by activating histone demethylase JMJD3. Activated JMJD3 demethylates histone 3 lysine 27 trimethylation from the promoter of SNAI1 gene and leads to expression of Snail which is a critical inducer of EMT.
Repressive histone modifications
EZH2 is a histone methyltransferase which silences genes by methylation of histone 3 lysine 27 at the promoter. In several cancers, EZH2 is overexpressed resulting in silencing of genes such as CDH1, AXIN2, NKD1, PP2R2B, which are required for either maintaining epithelial phenotype or controlling EMT inducers. When these genes are silenced, the cells undergo EMT, and the cancer becomes metastatic.
Snail (SNAI1) which is the master inducer of EMT has been found to recruit histone methyltransferase G9a to deposit repressive histone 3 lysine 9 methylation marks on the promoter of epithelial marker gene CDH1.
SNAI1 also recruits SIN3A complex to the epithelial marker gene CDH1. SIN3A complex contains histone deacetylases. As deacetylation of activating histone acetyl marks can repress genes, SIN3A can repress CDH1 and allow cells to undergo EMT.
Conclusion
Metastasis is a well-known factor associated with a worse prognosis in cancer. Malignant cells become metastatic by exploiting several biological processes, one of which is EMT. In many cancers, aberrations in histone post-translational modification leads to altered expression of key EMT genes, which increases the metastatic potential of malignant cells. Continuing research into both the process and the genes involved in histone modifications that lead to metastatic cancer has great potential to improve diagnosis and treatment.
Learn more:
Acknowledgements:
We would like to thank Dr. Linda Penn and Dr. Mohammadi Gharrari from Princess Margaret Cancer Centre for providing expert review of this article. Thank you also to Elizabeth Elder for providing the French translation.
Images created in www.BioRender.com
References:
1. Wittekind C, Neid M. Cancer Invasion and Metastasis. Oncology. 2005;69(Suppl. 1):14-16. doi:10.1159/000086626
2. Definition of carcinoma in situ - NCI Dictionary of Cancer Terms - NCI. Published February 2, 2011.
3. Lambert AW, Pattabiraman DR, Weinberg RA. Emerging Biological Principles of Metastasis. Cell. 2017;168(4):670-691. doi:10.1016/j.cell.2016.11.037
4. Divoli A, Mendonça EA, Evans JA, Rzhetsky A. Conflicting Biomedical Assumptions for Mathematical Modeling: The Case of Cancer Metastasis. PLOS Comput Biol. 2011;7(10):e1002132. doi:10.1371/journal.pcbi.1002132
5. Nguyen DX, Bos PD, Massagué J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9(4):274-284. doi:10.1038/nrc2622
6. Gros J, Tabin CJ. Vertebrate Limb bud formation is initiated by localized Epithelial to Mesenchymal Transition. Science. 2014;343(6176):1253-1256. doi:10.1126/science.1248228
7. Correa-Costa M, Andrade-Oliveira V, Braga TT, et al. Activation of platelet-activating factor receptor exacerbates renal inflammation and promotes fibrosis. Lab Invest. 2014;94(4):455-466. doi:10.1038/labinvest.2013.155
8. Banerjee P, Venkatachalam S, Mamidi MK, Bhonde R, Shankar K, Pal R. Vitiligo patient-derived keratinocytes exhibit characteristics of normal wound healing via epithelial to mesenchymal transition. Exp Dermatol. 2015;24(5):391-393. doi:10.1111/exd.12671
9. Grande MT, Sánchez-Laorden B, López-Blau C, et al. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat Med. 2015;21(9):989-997. doi:10.1038/nm.3901
10. Nakaya Y, Sukowati EW, Wu Y, Sheng G. RhoA and microtubule dynamics control cell–basement membrane interaction in EMT during gastrulation. Nat Cell Biol. 2008;10(7):765-775. doi:10.1038/ncb1739
11. Roche J. The Epithelial-to-Mesenchymal Transition in Cancer. Cancers. 2018;10(2):52. doi:10.3390/cancers10020052
12. Jolly MK, Mani SA, Levine H. Hybrid epithelial/mesenchymal phenotype(s): The ‘fittest’ for metastasis? Biochim Biophys Acta BBA - Rev Cancer. 2018;1870(2):151-157. doi:10.1016/j.bbcan.2018.07.001
13. Thiery JP. Epithelial–mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2(6):442-454. doi:10.1038/nrc822
14. Chiang SPH, Cabrera RM, Segall JE. Tumor cell intravasation. Am J Physiol - Cell Physiol. 2016;311(1):C1-C14. doi:10.1152/ajpcell.00238.2015
15. Strilic B, Offermanns S. Intravascular Survival and Extravasation of Tumor Cells. Cancer Cell. 2017;32(3):282-293. doi:10.1016/j.ccell.2017.07.001
16. McGhee JD, Felsenfeld G. Nucleosome Structure. Annu Rev Biochem. 1980;49(1):1115-1156. doi:10.1146/annurev.bi.49.070180.005343
17. Henikoff S, Furuyama T, Ahmad K. Histone variants, nucleosome assembly and epigenetic inheritance. Trends Genet. 2004;20(7):320-326. doi:10.1016/j.tig.2004.05.004
18. Jenuwein T, Allis CD. Translating the Histone Code. Science. 2001;293(5532):1074-1080. doi:10.1126/science.1063127
19. Bartee L, Shriner W, Creech C. Eukaryotic epigenetic regulation. Published online 2017.
20. Hake SB, Xiao A, Allis CD. Linking the epigenetic ‘language’ of covalent histone modifications to cancer. Br J Cancer. 2004;90(4):761-769. doi:10.1038/sj.bjc.6601575
21. Markouli M, Strepkos D, Basdra EK, Papavassiliou AG, Piperi C. Prominent Role of Histone Modifications in the Regulation of Tumor Metastasis. Int J Mol Sci. 2021;22(5):2778. doi:10.3390/ijms22052778
22. Wang JQ, Yan FQ, Wang LH, et al. Identification of new hypoxia-regulated epithelial-mesenchymal transition marker genes labeled by H3K4 acetylation. Genes Chromosomes Cancer. 2020;59(2):73-83. doi:10.1002/gcc.22802
23. Dhasarathy A, Phadke D, Mav D, Shah RR, Wade PA. The Transcription Factors Snail and Slug Activate the Transforming Growth Factor-Beta Signaling Pathway in Breast Cancer. PLOS ONE. 2011;6(10):e26514. doi:10.1371/journal.pone.0026514
24. Kapoor-Vazirani P, Kagey JD, Powell DR, Vertino PM. Role of hMOF-dependent histone H4 lysine 16 acetylation in the maintenance of TMS1/ASC gene activity. Cancer Res. 2008;68(16):6810-6821. doi:10.1158/0008-5472.CAN-08-0141
25. Zhao L, Li W, Zang W, et al. JMJD2B promotes epithelial-mesenchymal transition by cooperating with β-catenin and enhances gastric cancer metastasis. Clin Cancer Res Off J Am Assoc Cancer Res. 2013;19(23):6419-6429. doi:10.1158/1078-0432.CCR-13-0254
26. Usman S, Waseem NH, Nguyen TKN, et al. Vimentin Is at the Heart of Epithelial Mesenchymal Transition (EMT) Mediated Metastasis. Cancers. 2021;13(19):4985. doi:10.3390/cancers13194985
27. Björkman M, Östling P, Härmä V, et al. Systematic knockdown of epigenetic enzymes identifies a novel histone demethylase PHF8 overexpressed in prostate cancer with an impact on cell proliferation, migration and invasion. Oncogene. 2012;31(29):3444-3456. doi:10.1038/onc.2011.512
28. Ramadoss S, Chen X, Wang CY. Histone Demethylase KDM6B Promotes Epithelial-Mesenchymal Transition. J Biol Chem. 2012;287(53):44508-44517. doi:10.1074/jbc.M112.424903
29. Ferraro A, Mourtzoukou D, Kosmidou V, et al. EZH2 is regulated by ERK/AKT and targets integrin alpha2 gene to control Epithelial–Mesenchymal Transition and anoikis in colon cancer cells. Int J Biochem Cell Biol. 2013;45(2):243-254. doi:10.1016/j.biocel.2012.10.009
30. Dong C, Wu Y, Wang Y, et al. Interaction with Suv39H1 is critical for Snail-mediated E-cadherin repression in breast cancer. Oncogene. 2013;32(11):1351-1362. doi:10.1038/onc.2012.169
31. Peinado H, Ballestar E, Esteller M, Cano A. Snail mediates E-cadherin repression by the recruitment of the Sin3A/histone deacetylase 1 (HDAC1)/HDAC2 complex. Mol Cell Biol. 2004;24(1):306-319. doi:10.1128/MCB.24.1.306-319.2004