Plasticité épigénétique dans la transition du cancer de la prostate résistant à la castration (CPRC) vers le cancer de la prostate neuroendocrinien (CPNE)
par Shaghayegh Nouruzi
La progression du cancer de la prostate résistant à la castration (CRPC) vers le cancer neuroendocrinien de la prostate (NEPC) s'accompagne d'une plasticité épigénétique, d'un remodelage de la chromatine, de modifications des histones et d'une modification des modèles de méthylation de l'ADN. Des facteurs de transcription clés, tels que ASCL1, et des modificateurs de chromatine tels que EZH2 et DNMT, apparaissent comme des acteurs essentiels dans le développement de ce stade agressif de la maladie.
Plasticité épigénétique dans la transition du cancer de la prostate résistant à la castration (CPRC) vers le cancer de la prostate neuroendocrinien (CPNE)
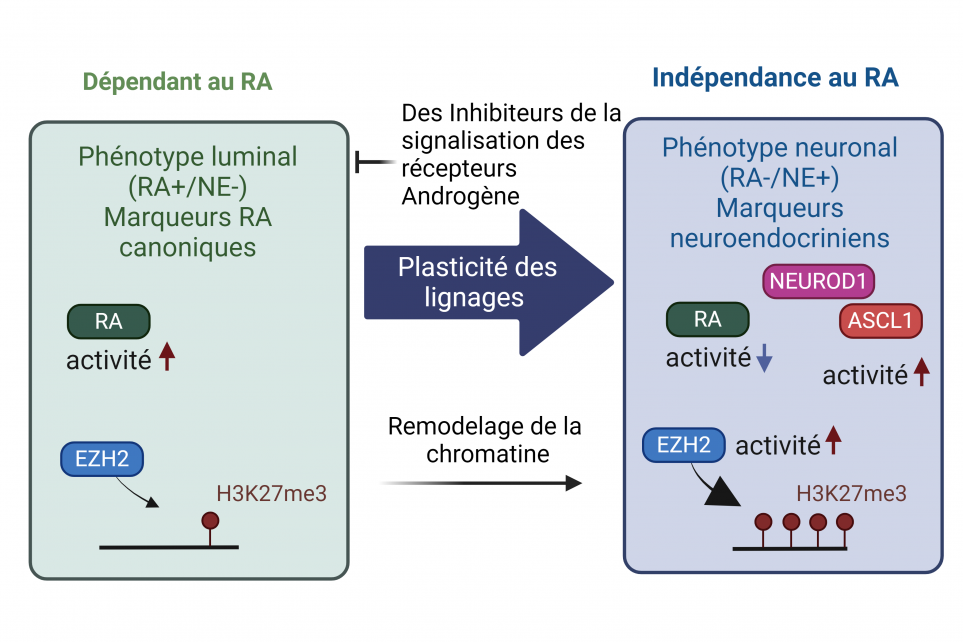
Malgré le succès significatif des Inhibiteurs de la signalisation des récepteurs androgene (ISRA) de seconde génération (tels que l'enzalutamide et l'abiratérone), les cancers de la prostate résistants à la castration (CPRC) parviennent à progresser malgré une privation androgénique prolongée. Un sous-ensemble de tumeurs CPRC échappe à la pression des thérapies ciblées en perdant leur dépendance au récepteur des androgènes (RA) (indépendance au RA). Ces nouveaux clones possèdent alors des profils moléculaires distincts et développent un nouveau phénotype associé à des caractéristiques de cellules souches et neuronales. La plasticité des clones sous-tend cette transition vers un phénotype alternatif. En effet, ce processus est initialement conduit par des altérations moléculaires qui créent des clones avec une plasticité cellulaire, suivies d'événements supplémentaires conduisant à l'émergence du cancer de la prostate neuroendocrinien à petites cellules induit par le traitement (tCPNE). La plupart des cas de CPNE sont diagnostiqués à des stades avancés, avec métastases, expliquant le taux élevé de mortalité. Bien que les cas de CPNE émergeant de novo soient rares, les tCPNE représentent environ 20 % des cas avancés et sont réfractaires aux traitements du CPRC. Ce chiffre pourrait augmenter en raison de l'introduction plus précoce des thérapies hormonales dans la ligne de traitement.
Le paysage épigénétique du CPNE
Les variations génomiques entre le CPRC et le CPNE sont limitées, à l'exception de la perte fréquente de suppresseurs de tumeurs tels que PTEN, RB1, et TP53. Cependant, la progression vers le CPNE est accompagnée d'une reprogrammation transcriptionnelle étendue, suggérant que l'émergence du phénotype neuroendocrinien est principalement due à une dérégulation épigénétique. La plasticité épigénétique contrôle le destin cellulaire en modifiant l'accessibilité de l'ADN aux facteurs de transcription et l'expression des gènes. Ce processus est régulé notamment par la méthylation de l'ADN, le remodelage des nucléosomes et les modifications des histones. Finalement, les altérations de l'épigénome des tumeurs CPRC après des thérapies ciblant le RA caractérisent la progression vers le tCPNE.
Remodelage de la chromatine et rôle des facteurs de transcription
La progression du CPRC vers le tCPNE sous la pression des ISRAs est marquée par un remodelage significatif de la chromatine, où l'accessibilité de la chromatine augmente dans les régions régulant les voies neuronales et de cellules souches, facilitant ainsi la liaison de facteurs de transcription spécifiques (FTs). Cela provoque un changement des profils d'expression des gènes vers un phénotype neuronal. Au cours de la progression vers le CPNE, ASCL1, un facteur de transcription déterminant neuronal, joue un rôle central en favorisant la plasticité des lignages et la détermination neuronale. L'expression d'ASCL1, ainsi que son accessibilité et son activité, augmentent de manière disproportionnée après en réponse aux ISRA, menant à l'activation de programmes neuronaux et de cellules souches. Notamment, la perte d'ASCL1 dans le CPNE entraîne une perte significative d'accessibilité chromatinienne et un retour à un état luminal. ASCL1 soutient également le phénotype CPNE en reprogrammant le cistrome d'autres FTs pionniers comme FOXA1. Ainsi, la transition du CPRC au CPNE induite par les ISRA modifie le contrôle transcriptionnel des facteurs de transcription spécifiques aux lignages.
Il est important de souligner la distinction entre les sous-types de CPNE à travers leur paysage chromatinien, car chaque sous-type possède des vulnérabilités uniques qui peuvent être exploitées à des fins thérapeutiques. Bien que convergeant vers un programme neuronal, des facteurs de transcription déterminants de la lignée neuronale tels qu'ASCL1 et NEUROD1 orchestrent des trajectoires de remodelage chromatinien divergentes qui favorisent le programme neuroendocrinien (NE). En outre, des études utilisant des analyses d'accessibilité chromatinienne à cellule unique chez des modèles murins ont révélé deux clones distincts, conduits par ASCL1 ou par le facteur de transcription spécifique aux cellules souches OCT11 (également connu sous le nom de POU2F3), définit un sous-type non-NE. Ces résultats mettent en lumière la complexité du paysage épigénétique dans le CPNE et la spécificité du rôle des FTs dans le développement du phénotype NE.

Modificateurs épigénétiques
Les facteurs de transcription déterminant les lignages guident des modificateurs spécifiques de la chromatine et épigénétiques pour établir le destin cellulaire souhaité. Les modificateurs épigénétiques régissant les changements de lignées dans le cancer de la prostate sont souvent dérégulés dans le CPNE, tels que la méthyltransférase des histones EZH2 et les ADN méthyltransférases. EZH2, un composant clé du complexe Polycomb Répressif 2 (PRC2), dépose des groupes méthyle sur la lysine 27 de l'histone 3 (H3K27) pour réprimer la transcription. EZH2 est surexprimé dans le CPNE par rapport au CPRC et joue un rôle dans la suppression du programme luminal dépendant au RA. De manière intéressante, il a été rapporté qu'EZH2 adopte un fonctionnement non-canonique, en concert avec un programme alternatif au RA, pour induire l'acétylation des histones et faciliter la conversion de la lignée NE afin d'activer les programmes de cellules souches et de NE.
Parallèlement, le rôle de la méthylation de l'ADN dans la distinction des stades du cancer de la prostate devient de plus en plus évident. Les altérations du méthylome de l'ADN sont une caractéristique du tCPNE, et participent au profil épigénétique unique de ces tumeurs. En effet, un profil distinct de méthylation de l'ADN (combinaison d'hyperméthylation et d'hypométhylation de l'ADN) est associé au CPNE et peut être détecté dans l'ADN circulant libre (cfDNA), offrant ainsi des biomarqueurs potentiellement significatifs. Cependant, il reste à déterminer si les sous-types de CPNE peuvent être distingués par des profils de méthylation de l'ADN différents. Ces interactions complexes entre les FTs déterminant les lignées et les modificateurs épigénétiques soulignent la complexité de la progression du cancer de la prostate et le développement de la plasticité des lignées. Étant donné le potentiel de réversibilité des modifications épigénétiques, le décryptage des mécanismes contrôlant la plasticité des lignées pourrait conduire à l'identification de cibles thérapeutiques et au développement de stratégies pour combattre la résistance aux ISRAs.
Apprendre encore plus:
Ces articles de revue résument les mécanismes transcriptionnels et épigénétiques régissant la plasticité de la lignée du cancer de la prostate :
- The Transcriptional and Epigenetic Landscape of Cancer Cell Lineage Plasticity | Cancer Discovery | American Association for Cancer Research (aacrjournals.org)
- Epigenetics in prostate cancer: clinical implications - PMC (nih.gov)
- Epigenetic reprogramming during prostate cancer progression: A perspective from development - PMC (nih.gov)
Images créées sur www.BioRender.com
References:
- Scher, H.I. et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med367, 1187-1197 (2012).
- de Bono, J.S. et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med 364, 1995-2005 (2011).
- Kaarijarvi, R., Kaljunen, H. & Ketola, K. Molecular and Functional Links between Neurodevelopmental Processes and Treatment-Induced Neuroendocrine Plasticity in Prostate Cancer Progression. Cancers (Basel)13 (2021).
- Vlachostergios, P.J., Puca, L. & Beltran, H. Emerging Variants of Castration-Resistant Prostate Cancer. Curr Oncol Rep 19, 32 (2017).
- Aggarwal, R. et al. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J Clin Oncol 36, 2492-2503 (2018).
- Beltran, H. et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov 1, 487-495 (2011).
- Aparicio, A., Logothetis, C.J. & Maity, S.N. Understanding the lethal variant of prostate cancer: power of examining extremes. Cancer Discov 1, 466-468 (2011).
- Zou, M. et al. Transdifferentiation as a Mechanism of Treatment Resistance in a Mouse Model of Castration-Resistant Prostate Cancer. Cancer Discov 7, 736-749 (2017).
- Ellis, L. & Loda, M. Advanced neuroendocrine prostate tumors regress to stemness. Proc Natl Acad Sci U S A112, 14406-14407 (2015).
- Crona, D.J. & Whang, Y.E. Androgen Receptor-Dependent and -Independent Mechanisms Involved in Prostate Cancer Therapy Resistance. Cancers (Basel) 9 (2017).
- Beltran, H. et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med 22, 298-305 (2016).
- Abdulfatah, E. et al. De novo neuroendocrine transdifferentiation in primary prostate cancer-a phenotype associated with advanced clinico-pathologic features and aggressive outcome. Med Oncol 38, 26 (2021).
- Wang, H.T. et al. Neuroendocrine Prostate Cancer (NEPC) progressing from conventional prostatic adenocarcinoma: factors associated with time to development of NEPC and survival from NEPC diagnosis-a systematic review and pooled analysis. J Clin Oncol 32, 3383-3390 (2014).
- Beltran, H. et al. Impact of Therapy on Genomics and Transcriptomics in High-Risk Prostate Cancer Treated with Neoadjuvant Docetaxel and Androgen Deprivation Therapy. Clin Cancer Res 23, 6802-6811 (2017).
- Labrecque, M.P. et al. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J Clin Invest 129, 4492-4505 (2019)
- Helpap, B., Kollermann, J. & Oehler, U. Neuroendocrine differentiation in prostatic carcinomas: histogenesis, biology, clinical relevance, and future therapeutical perspectives. Urol Int 62, 133-138 (1999).
- Zaffuto, E. et al. Contemporary Incidence and Cancer Control Outcomes of Primary Neuroendocrine Prostate Cancer: A SEER Database Analysis. Clin Genitourin Cancer 15, e793-e800 (2017).
- Epstein, J.I. et al. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. Am J Surg Pathol 38, 756-767 (2014)
- Linder, S. et al. Drug-Induced Epigenomic Plasticity Reprograms Circadian Rhythm Regulation to Drive Prostate Cancer toward Androgen Independence. Cancer Discov 12, 2074-2097 (2022).
- Davies, A., Zoubeidi, A. & Selth, L.A. The epigenetic and transcriptional landscape of neuroendocrine prostate cancer. Endocr Relat Cancer 27, R35-R50 (2020).
- Chakraborty, G., Gupta, K. & Kyprianou, N. Epigenetic mechanisms underlying subtype heterogeneity and tumor recurrence in prostate cancer. Nat Commun 14, 567 (2023).
- Nouruzi, S. et al. ASCL1 activates neuronal stem cell-like lineage programming through remodeling of the chromatin landscape in prostate cancer. Nat Commun 13, 2282 (2022).
- Davies, A. et al. An androgen receptor switch underlies lineage infidelity in treatment-resistant prostate cancer. Nat Cell Biol 23, 1023-1034 (2021).
- Bauer, A.J. & Martin, K.A. Coordinating Regulation of Gene Expression in Cardiovascular Disease: Interactions between Chromatin Modifiers and Transcription Factors. Front Cardiovasc Med 4, 19 (2017).
- Baca, S.C. et al. Reprogramming of the FOXA1 cistrome in treatment-emergent neuroendocrine prostate cancer. Nat Commun 12, 1979 (2021).
- Ku, S.Y. et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355, 78-83 (2017)
- Smith, B.A. et al. A Human Adult Stem Cell Signature Marks Aggressive Variants across Epithelial Cancers. Cell Rep 24, 3353-3366 e3355 (2018).
- Akamatsu, S., Inoue, T., Ogawa, O. & Gleave, M.E. Clinical and molecular features of treatment-related neuroendocrine prostate cancer. Int J Urol 25, 345-351 (2018).
- Beltran, H. et al. Circulating tumor DNA profile recognizes transformation to castration-resistant neuroendocrine prostate cancer. J Clin Invest 130, 1653-1668 (2020).
- Berchuck, J.E. et al. Detecting Neuroendocrine Prostate Cancer Through Tissue-Informed Cell-Free DNA Methylation Analysis. Clin Cancer Res 28, 928-938 (2022).