A sensitive analytical tool to study epigenetic modifications and protein-DNA interactions from ChIP-Seq data.
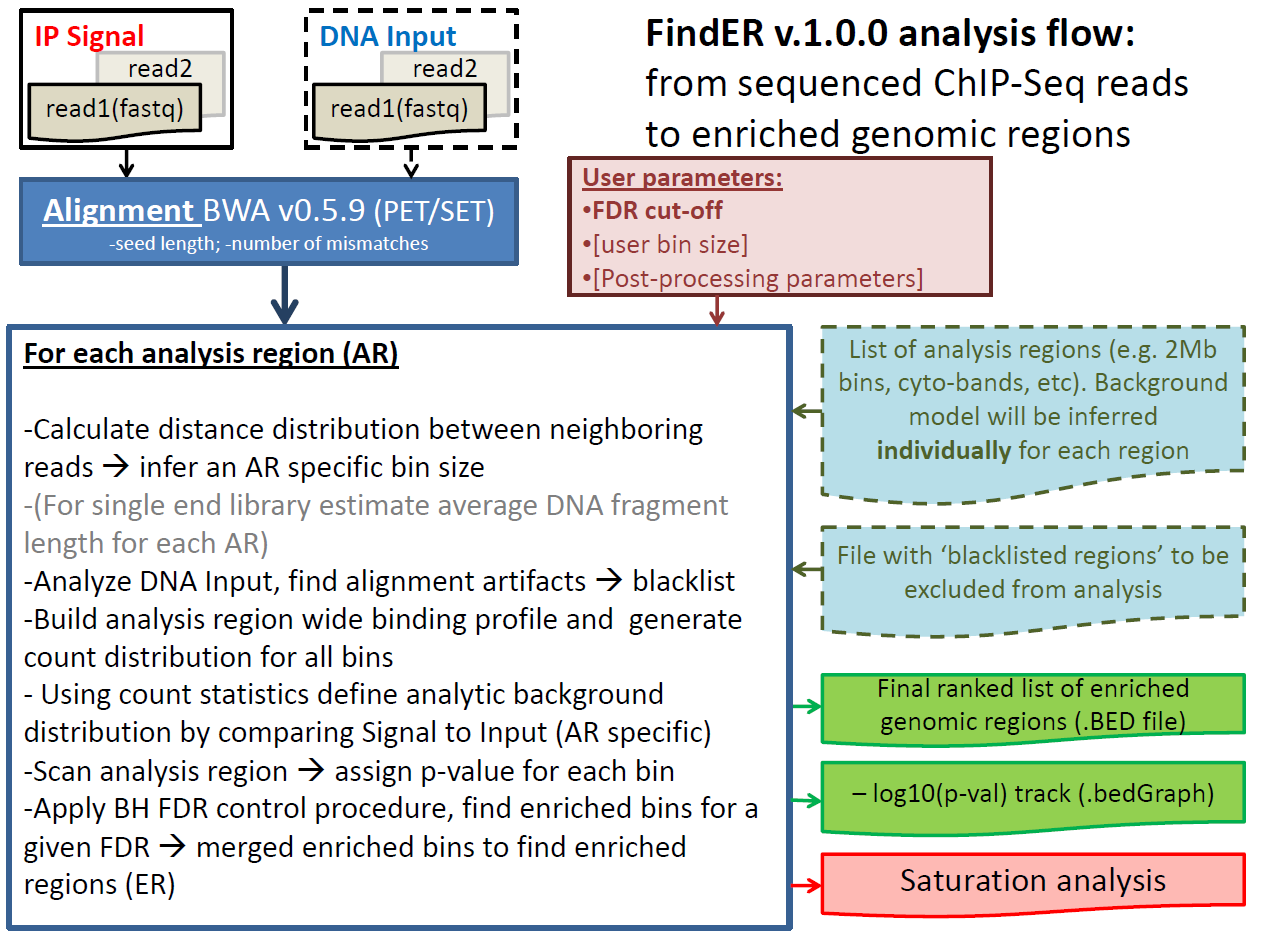
We present a versatile analysis tool developed to Find Enriched Regions (FindER v1.0.0) in ChIP-Seq datasets. FindER is intended to overcome sequence depth limitations of many existing tools and provides a common mechanism for identifying enrichment from localized (e.g. H3K4me3 histone modification, or DNA-protein binding) as well as dispersed (e.g. H3K27me3, H3K36me3) ChIP-Seq signal profiles, or mixed of two signal types (e.g. H3K4me1) . After reading aligned IP-signal data and DNA Input control data in the BAM format, FindER computationally segments genome into bins of size defined by the read density distribution in the IP data. Using these adaptive bins FindER first blacklists alignment artifacts (that are typically appear as spurious enriched regions) using Input DNA control (with an option to use external list of blacklisted regions as well). FindER then compares count statistics for the IP signal vs. control to obtain a threshold separating the genome into background and enriched bins. This approach is independent of the unknown relative normalization between IP and control data sets. After that the proportion of the background like reads in the IP-data is evaluate, control data is rescaled and the significance of the enriched bins is then refined with a False Discovery Rate control process. This signal-background separation process can be applied globally or locally. It is free from assumptions about underlying distributions of read density and therefore enables the generation of a normalized signal profile after taking into account the local signal-to-noise ratio [currently, -log(p-value)]. This allows for integrative multi-sample analysis for data with comparable signal-to-noise ratio, and is especially important in examining the effect of relative signal strength on the biology. Additionally various metrics that can be used to evaluate a success of the experiment such as signal to noise ratio are also computed. Number of post-processing options for combining co-located enriched regions and filtering on region size is embedded into the tool. The saturation study using different subsampling strategies is available to the user. User can also chose to use FindER to rank significance of enrichment of the IP-signal in a set of selected regions (e.g. gene promoters, known TF binding sites, etc). Crucially, FindER is a production grade application. It is a user friendly tool implemented in Java, and it has been tested on terabyte scale ChIP-Seq data.
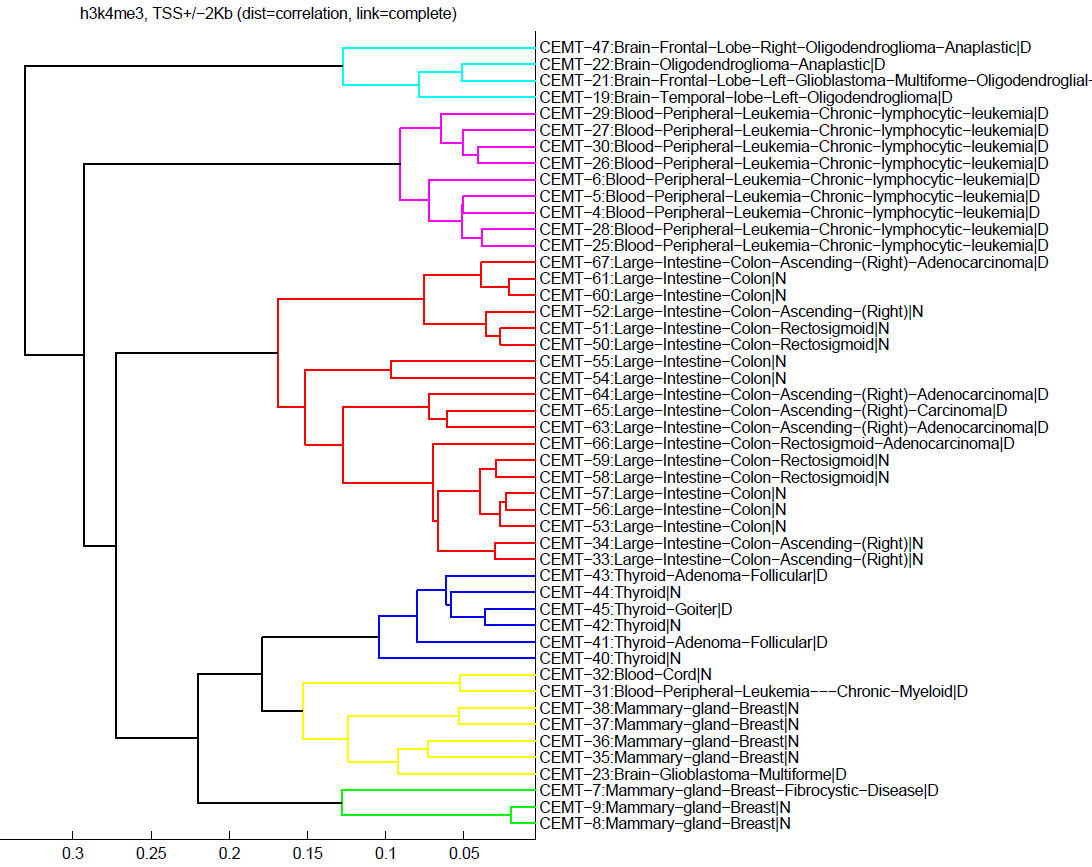
Here is a clustering example of a degree of enrichment found by FindER in the promoter regions (TSS+/-2Kb) of the coding genes for 48 CEMT samples.
Download and usage
FindER is available as a JAR file: FindER.1.0.0e.jar.
The corresponding md5sum is: FindER.1.0.0e.jar.md5sum .
In order to run FindER you have to have java 7 installed (please see Oracle website for details).
FindER uses samtools-0.1.x [Note program won't work correctly with samtools-1.x]. If you don't have samtools-0.1.x installed, please download it from samtools-0.1.17 website.
To see on-line help, please use:
java -jar FindER.jar -h
command.
Currently typical FindER run requires ~10G of memory (-Xmx10G).
Updates log
13/11/2015: FindER v 1.0.0 beta is released.
18/07/2016: Bug preventing FindER v 1.0.0 beta to run on hg38 alignments fixed